
The Rare Atlas
This page provides an overview of the many ways rare diseases can be classified. The tables include representative examples for each subcategory, supporting identification of underserved disease areas and potential gaps in therapeutic development.
Age of Onset
| Onset | Examples | Clinical Relevance |
|---|---|---|
| Congenital (Neonatal) | Spinal Muscular Atrophy Type 1 Tay–Sachs disease MPS I (Hurler syndrome) Alström syndrome Cerebral creatine deficiency | Diagnosed at or shortly after birth; Often fatal without early intervention |
| Childhood-Onset | Duchenne Muscular Dystrophy Batten disease Rett syndrome Pompe disease Sanfilippo syndrome | Often progressive; Major impact on families; Many lack disease-modifying therapies |
| Adolescent-Onset | Wilson’s disease Friedreich’s ataxia X-linked adrenoleukodystrophy Erythromelalgia Glycogen storage disease type III | Early symptoms often missed or misdiagnosed; Limited transition care from paediatric to adult |
| Adult-Onset | Amyloidosis Pulmonary alveolar proteinosis Huntington’s disease CADASIL Hereditary angioedema | Delayed diagnosis; Some mimic common diseases; Ethical concerns in predictive testing |
| Late-Onset (Geriatric) | Niemann–Pick type C (adult form) Multiple system atrophy Late-onset Pompe Fabry (cardiac variant) Inclusion body myositis | Often misdiagnosed as dementia or age-related disorders; Missed therapeutic window |
Organs
| Organ | Examples | Clinical Relevance |
|---|---|---|
| Neurological | Gerstmann–Sträussler–Scheinker syndrome Chorea-acanthocytosis Huntington’s disease CADASIL CLN11 | Ultra-rare or dominantly inherited neurodegeneration with little or no treatment |
| Ophthalmological | Leber congenital amaurosis Retinitis pigmentosa Aniridia Stargardt disease Choroideremia | Rare inherited retinal diseases; Targets of gene therapy trials |
| Auditory | Usher syndrome Pendred syndrome Waardenburg syndrome Otosclerosis Connexin 26-related hearing loss | Hearing loss or deafness, sometimes combined with balance or vision issues; Limited curative therapies; Early intervention improves quality of life |
Cardiovascular | Fabry disease (cardiac form) Danon disease Barth syndrome PRKAG2 cardiomyopathy Carvajal syndrome | Often misdiagnosed as common cardiomyopathies; Serious but under-recognized |
| Gastrointestinal | Hirschsprung disease Celiac disease (rare variants) Short bowel syndrome Familial adenomatous polyposis Progressive familial intrahepatic cholestasis | Affects digestion, nutrient absorption, and liver function; Treatment varies from dietary management to surgery; Can severely impact growth and quality of life |
| Renal | Alport syndrome Polycystic kidney disease Nephrotic syndrome (congenital forms) Fabry disease Cystinosis | Causes kidney dysfunction, proteinuria, and progressive renal failure; Dialysis or transplantation may be required; Limited disease-modifying therapies |
| Skeletal | Fibrodysplasia ossificans progressiva Osteogenesis imperfecta Cleidocranial dysplasia Hypophosphatasia Campomelic dysplasia | Many have no approved therapies and high physical burden |
| Dermatological | Epidermolysis bullosa Ichthyosis vulgaris Xeroderma pigmentosum Darier disease Netherton syndrome | Chronic skin fragility, scaling, or cancer risk; Significant impact on quality of life; Management mainly supportive and protective |
System
| System | Examples | Clinical Relevance |
|---|---|---|
| Metabolic | PMM2-CDG PGM1-CDG MPS I (Hurler) Metachromatic leukodystrophy Niemann–Pick disease type C | Involve enzyme deficiencies; Some with enzyme or gene therapies, many without |
| Respiratory | Cystic fibrosis Primary ciliary dyskinesia Alpha-1 antitrypsin deficiency Surfactant metabolism disorders Pulmonary alveolar proteinosis | Impaired lung function, recurrent infections, and progressive respiratory failure; Treatments range from supportive care to lung transplantation |
| Neurological | Rett syndrome CLN3 Batten disease Joubert syndrome Friedreich’s ataxia X-linked adrenoleukodystrophy | Neurodegenerative/ Neurodevelopmental/ Mixed presentations |
| Immunological | Chronic granulomatous disease Hyper IgM syndrome DOCK8 deficiency SCID WHIM syndrome | Serious early-onset diseases; Many under registry surveillance, limited access to care |
| Haematological | Fanconi anemia Diamond–Blackfan anemia Paroxysmal nocturnal hemoglobinuria Chronic granulomatous disease overlaps Rare coagulopathies | Blood component dysfunctions; Risk of overlooked morbidity |
| Mitochondrial | Leigh syndrome MELAS Alström syndrome MERRF POLG-related disorders | Mitochondrial dysfunction causes multi-organ failure; Limited therapies, poor survival |
| Endocrine | Congenital adrenal hyperplasia Hypopituitarism MEN1 syndrome Wolfram syndrome Diabetes insipidus | Hormonal imbalances affecting growth, metabolism, and homeostasis; Treatment involves hormone replacement or targeted therapies; Lifelong monitoring needed |
| Urinary & Reproductive | Lowe syndrome Alport syndrome ARPKD Rare congenital tract malformations Rare tumours | Underdiagnosed combinations of renal and reproductive systems |
Others
| Category | Examples | Clinical Relevance |
|---|---|---|
| Congenital | Spina bifida Apert syndrome Limb-body wall complex Sirenomelia Gastroschisis | Visible at birth; Often multisystem, critical for early research and registry |
| Cancer | Retinoblastoma Neuroblastoma Li-Fraumeni syndrome-associated cancers Medullary thyroid carcinoma Chronic myeloid leukemia (rare variants) | Malignant growths with variable prognosis; Treatment may include surgery, chemotherapy, targeted therapy; Early diagnosis improves survival |
| Infectious | Congenital cytomegalovirus infection Chronic hepatitis B (rare cases) HIV-related opportunistic infections Primary immunodeficiency-associated infections Tuberculosis (rare pediatric forms) | Infects specific organs or systems, often with chronic or life-threatening consequences; Treatments include antivirals, antibiotics, or supportive care; Prevention and early treatment are critical |
| Neurological | Rett syndrome Joubert syndrome Huntington’s Gerstmann–Sträussler syndrome Friedreich’s ataxia | Often progressive; High burden; Therapeutic gaps |
| Genetic | NGLY1 deficiency CDKL5 deficiency TBL1XR1 disorder KAT6A syndrome Rett syndrome | Affect transcriptional or epigenetic regulation; Very rare, often syndromic |
| Metabolic | Gaucher disease Pompe disease PMM2-CDG ALG1-CDG Niemann–Pick disease type C | Enzyme disorders often detectable early but many remain underserved |
Research Status
| Status | Examples | Clinical Relevance |
|---|---|---|
| No Approved Therapy | NGLY1 Deficiency Fibrodysplasia ossificans progressiva CHAMP1 disorder Ullrich congenital muscular dystrophy Joubert syndrome | No approved drugs; Families rely on off-label use or repurposing |
| Preclinical (Early R&D) | Rare splicing disorders CDKL5 deficiency CLN3-Batten Rare ciliopathies KAT6A syndrome | Research mostly in early academic phase; Few trials or commercial interest |
| In Clinical Trials | Leber congenital amaurosis Epidermolysis bullosa Methylmalonic acidemia Menkes disease Hereditary transthyretin amyloidosis | Trials underway, often with novel modalities (e.g., gene therapy, antisense oligos) |
| Approved Orphan Drug | Spinal Muscular Atrophy Cystic Fibrosis Pompe disease Fabry disease Gaucher disease | Have at least one approved drug; Access often country-dependent; Cost barriers persist |
| Repurposed Treatment | Propranolol for Hemangioma Sirolimus for lymphangioleiomyomatosis Miglustat for Niemann-Pick C Colchicine for FMF Sodium phenylbutyrate for UCDs | Off-label or new use of existing drugs; Sometimes only option available |
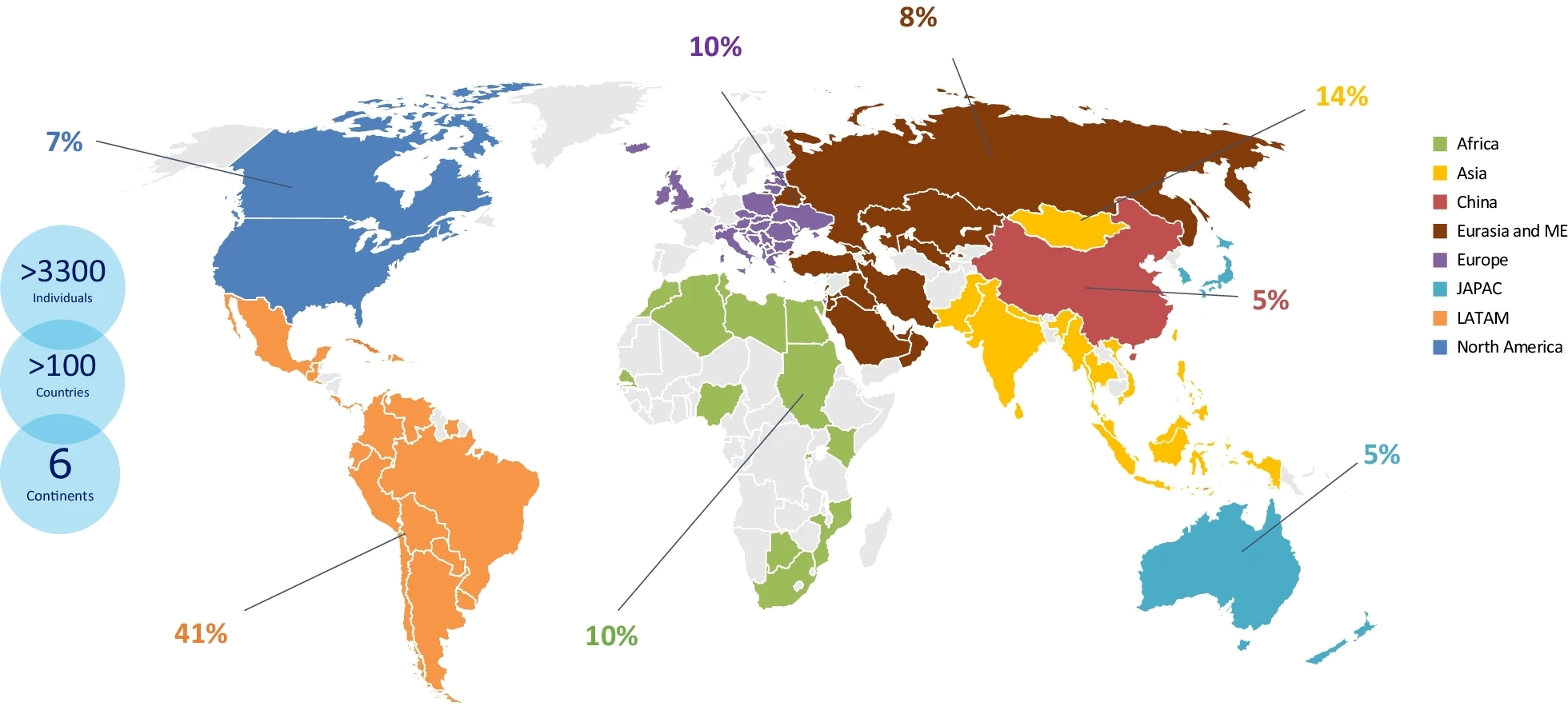
Percentage of individuals enrolled into the Humanitarian Program by region historically
Further Reading
Braun, I., Hartley, E., Olson, D., Matentzoglu, N., Schaper, K., Walls, R. and Vasilevsky, N., 2024. Increased discoverability of rare disease datasets through knowledge graph integration. JAMIA open, 8(1).
Cao, L., Sun, J. and Cross, A., 2024. An Automatic and End-to-End System for Rare Disease Knowledge Graph Construction Based on Ontology-Enhanced Large Language Models: Development Study. JMIR Medical Informatics, 12(1), p.e60665.
Chaudhary, A. and Kumar, V., 2025. Rare diseases: a comprehensive literature review and future directions. Journal of Rare Diseases, 4(1), p.33.
Debnath, A., Mazumder, R., Mazumder, A., Tyagi, P.K. and Singh, R.K., Challenges and Progress of Orphan Drug Development for Rare Diseases. Current pharmaceutical biotechnology.
EURORDIS‑Rare Diseases Europe, 2025. Rare Diseases Europe in 2025. [Accessed 6 Aug. 2025].
Evaluate Ltd., 2025. Orphan Drugs 2025: Are Orphans That Different? [Accessed 6 Aug. 2025].
Hivert, V., Jonker, A.H., O’Connor, D. and Ardigo, D., 2021. IRDiRC: 1000 new rare diseases treatments by 2027, identifying and bringing forward strategic actions. Rare Disease and Orphan Drugs Journal, 1(3).
Impact Global Health, 2025. The 2025 Neglected Disease R&D Pipeline Review: Time to Close the Gaps.[Accessed 6 Aug. 2025].
Loskill, P., Hardwick, R.N. and Roth, A., 2021. Challenging the pipeline. Stem Cell Reports, 16(9), pp.2033-2037.
National Center for Advancing Translational Sciences (NCATS). (n.d.). Genetic and Rare Diseases Information Center (GARD). National Institutes of Health. [Accessed 6 Aug. 2025].
National Center for Advancing Translational Sciences (NCATS), 2024. Rare Diseases Registry Program (RaDaR). [Accessed 1 October 2025].
Ogbogu, U. and Nel, A., 2024. Advanced Regenerative Medicines for Rare Diseases: A Review of Industry Sponsors Investment Motivations. Therapeutic innovation & regulatory science, 58(6), pp.1190-1199.
ResearchAndMarkets / GlobeNewswire, 2024. Gene Therapy in Rare Diseases – Therapeutics Pipeline Report 2024: Analytical Perspective… [Accessed 6 Aug. 2025].
Sanjak, J., Binder, J., Yadaw, A.S., Zhu, Q. and Mathé, E.A., 2024. Clustering rare diseases within an ontology-enriched knowledge graph. Journal of the American Medical Informatics Association, 31(1), pp.154-164.
Saviano, M., Barile, S., Caputo, F., Lettieri, M. and Zanda, S., 2019. From rare to neglected diseases: a sustainable and inclusive healthcare perspective for reframing the orphan drugs issue. Sustainability, 11(5), p.1289.
Whitcher, K., 2025. Rare Disease Day 2025: addressing the research divide. Research Communities, Springer Nature.[Accessed 6 Aug. 2025].
Zhu, Q., Nguyen, D.T., Grishagin, I., Southall, N., Sid, E. and Pariser, A., 2020. An integrative knowledge graph for rare diseases, derived from the Genetic and Rare Diseases Information Center (GARD). Journal of biomedical semantics, 11(1), p.13.
Zhu, Q., Qu, C., Liu, R., Vatas, G., Clough, A., Nguyễn, Ð.T., Sid, E., Mathé, E. and Xu, Y., 2022. Rare disease-based scientific annotation knowledge graph. Frontiers in Artificial Intelligence, 5, p.932665.
Categories may vary by classification, feel free to reach out if you think one is missing!
